
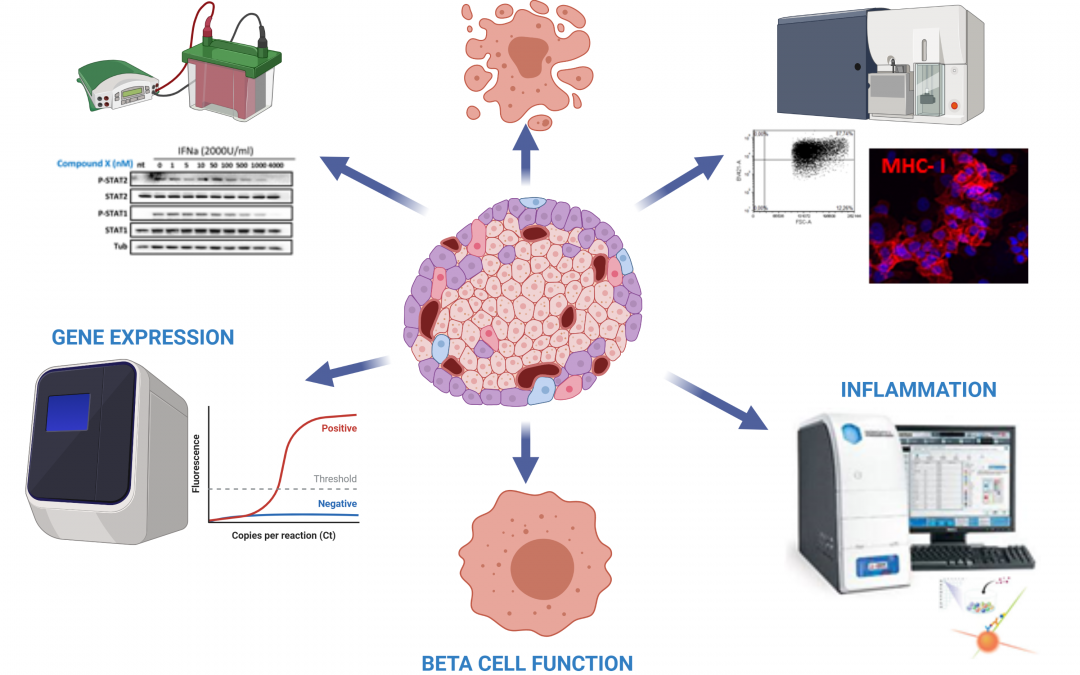
T
Type 1 diabetes (T1D) is a chronic autoimmune disease characterized by pancreatic islet inflammation and specific destruction of insulin-producing beta-cells by the immune system. Intriguingly, neighbouring glucagon-secreting alpha-cells survive the autoimmune attack, but are largely dysfunctional. During early phases of T1D, invading immune cells release several chemokines and cytokines into the islet milieu. Among these cytokines, interferon-alpha (IFNalpha), a member of the type I interferon (IFN) family, was observed in islets from T1D patients. Type I IFNs are critical for both innate and adaptive immune responses and may act as key links between genetic risk factors and environmental triggers in T1D pathogenesis. Despite the evidence suggesting that type I IFNs play a key role in early T1D, the molecular mechanisms underlying IFNalpha effects directly on pancreatic beta-cells are largely unknown. Our group has shown that IFNalpha is a common mediator of beta-cell MHC class I overexpression, endoplasmic reticulum stress and apoptosis, three hallmarks of early human T1D. IFNalpha also induced early modifications in chromatin remodelling and promoted major alterations in the alternative splicing landscape. Despite the knowledge that has been gathered about the effects of IFNalpha on beta-cells, much less is known about the mechanisms regulating type I IFN production and function in α-cells. It has been recently reported that alpha-cells expressed the highest levels of HLA class I in islets of autoantibody-positive donors as well as T1D patients. Moreover, alpha-cells expressed higher CXCL10 levels than beta-cells in pancreas of T1D subjects, indicating that alpha-cells may be the main contributors to the islet expression of CXCL10 in T1D. Thus, considering that type I IFN signalling is often cell-specific and the potential relevance of IFNalpha to T1D pathogenesis, it is crucial to define the global impact of IFNalpha on alpha-cells and characterize the differences between alpha- and beta-cells in response to this cytokine. Due to their role as key initiators of T1D pathogenesis, targeting type I IFN signalling pathway has arisen as a promising therapeutic approach to treat either at-risk individuals or patients in the very early stages of T1D. Inhibitors of Janus kinase (JAK) proteins, also known as JAKinibs, are among the strategies that present great potential. Recent reports show that clinically used JAKinibs prevented IFNalpha-induced signalling in human beta-cells, suggesting that they could be repurposed to treat T1D. However, as the current mode of action of first-generation JAKinibs present some specificity issues, efforts to discover new ways to inhibit these proteins are needed. Here we hypothesize that TYK2, a member of the JAK family of proteins, represents a good therapeutic target to prevent IFNalpha-induced effects on alpha- and beta-cells. Specifically, we used bioinformatics analyses to select two small molecules targeting the TYK2 FERM domain, which plays a key role in the interaction between TYK2 and the type I IFN receptor. The objectives of this project are:
1) To evaluate the global impact of IFNalpha on the transcriptional landscape of primary alpha- and beta-cells
2) To assess the effects of IFNalpha exposure on alpha- and beta-cell function
3) To assess whether the selected small molecules inhibit TYK2-mediated IFNalpha signalling pathway in alpha- and beta-cells